
计算Gene co-expression features
1614 人阅读 | 时间:2019年01月03日 22:03
Gene co-expression features
下载 co-expression 数据
The following co-expression coefficient features were attained from COXPRESdb.
http://coxpresdb.jp/download.shtml
打开这个页面我们点击bulk download


然后我们下载budding yeast 文件。
在最下面我们也可以看到文件格式的说明
Under the directory named Hsa.coex.v6, 19777 files will appear.
Hsa.coex.v6 ----- 1 |-- 10 |-- 100 |-- ... |-- 9997
1
462 8.1 0.596
2158 10.9 0.590
189 12.7 0.574
...
220963 19749.5 -0.163
130367 19760.5 -0.175 | 10
80168 4.9 0.553
10223 5.8 0.650
27284 5.9 0.608
...
84058 19772.0 -0.276
83871 19775.5 -0.304 | 100
85449 37.9 0.478
140807 47.7 0.391
636 50.2 0.469
...
126969 19269.8 -0.113
55930 19273.0 -0.082 |
Column 1; Entrez Gene ID of an opposite gene of coexpression (19776 genes)
Column 2; MR (Mutual Rank) as a final measure of coexpression. Lines are sorted by this value.
Column 3; Pearson‘s correlation coefficient of gene expression pattern
下载后的数据如下图
Sce.v14-08.G4461-S3819.rma.mrgeo.d文件夹包含4461个文件
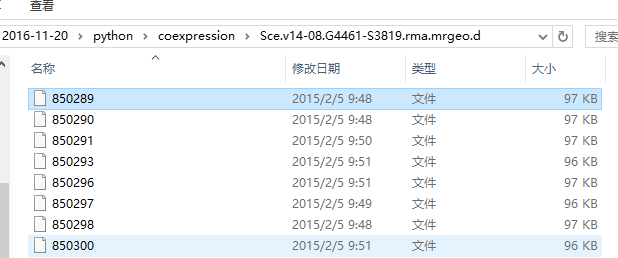
每个文件含有4461行对应4461个GENE ID
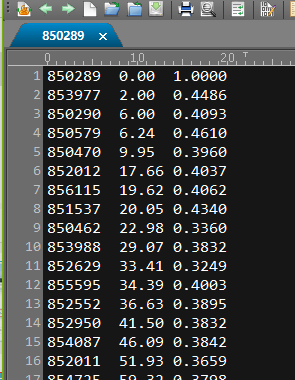
下载uniport数据
和前面同样一个问题,它这数据只给了3列,第一列 Entrez Gene ID,第二列MR,第三列COR。
蛋白质和 Entrez Gene ID的映射我们怎么得到呢?
通过各种资料。我们知道Entrez Gene ID就是ncbi里的ID,同样我们可以通过uniport下载对应的GENEID.
我们可以通过uniport上右边的笔头添加Gene ID列如下图所示
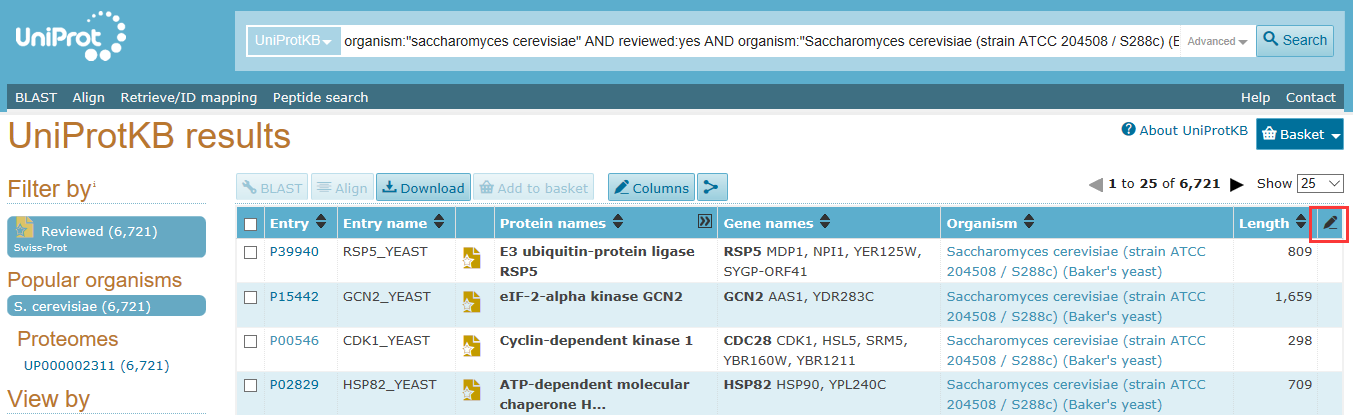
之后我们找到Genome annotation 下面的GeneID,打上勾(藏那么深也逃脱不了我的法眼。 )。这样我们就能得到GeneID信息了。
)。这样我们就能得到GeneID信息了。
)。这样我们就能得到GeneID信息了。
然后我们把下面的数据下载下来。

到此数据准备工作基本完成。
我们得到一个文件夹和一个uniprot和geneid对应关系的文件。
uniprot_to_geneid.csv
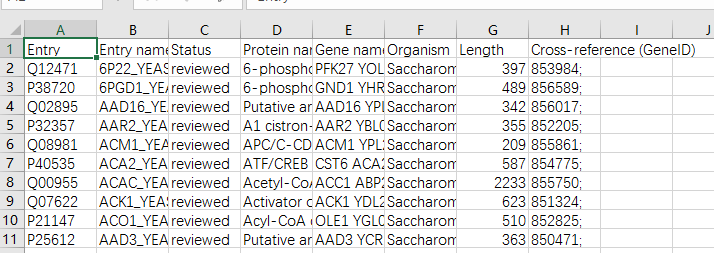
Ensemble learning prediction of protein–protein interactions using proteins functional annotations
获取论文的uniprot codes列(idA,idB)
yeast_gold_protein_pair.csv
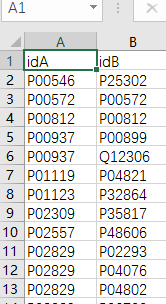
到这里我们的数据已经准备完成了。下面开始写代码!
代码的设计
# -*- coding: utf-8 -*-
"""
Created on Thu Nov 10 10:49:21 2016
@author: sun
"""
import pandas as pd
import os
yeast_gold_protein_pair=pd.read_csv(‘yeast_gold_protein_pair.csv‘,usecols=[‘idA‘,‘idB‘])
GeneID=pd.read_csv(‘uniprot_to_geneid.csv‘,usecols=[‘Entry‘,‘Cross-reference (GeneID)‘],index_col=0)
#注loc通过标签选择数据,iloc通过位置选择数据
idA=GeneID.loc[yeast_gold_protein_pair.idA,:]
idB=GeneID.loc[yeast_gold_protein_pair.idB,:]
idA.index=range(len(idA))
idB.index=range(len(idB))
mr=[]
cor=[]
for i in range(len(idA)):
GeneIDA=str(idA.iloc[i].values)
GeneIDB=str(idB.iloc[i].values)
ifGeneIDB!=‘[nan]‘andGeneIDA!=‘[nan]‘:
GeneIDA=GeneIDA[2:8]
GeneIDB=int(GeneIDB[2:8])
path=‘Sce.v14-08.G4461-S3819.rma.mrgeo.d/‘+GeneIDA
if os.path.exists(path):
coex=pd.read_csv(path,header=None,sep=‘ ‘,index_col=0)
ifGeneIDBin coex.index:
mr.append(coex.loc[GeneIDB,1])
cor.append(coex.loc[GeneIDB,2])
else:
mr.append("nan")
cor.append("nan")
else:
mr.append("nan")
cor.append("nan")
else:
mr.append("nan")
cor.append("nan")
yeast_gold_protein_pair[‘MR‘]=mr
yeast_gold_protein_pair[‘COR‘]=cor
yeast_gold_protein_pair.to_csv(‘coexpression.csv‘,index=False)最后我们可以得到一个coexpression.csv的文件,通过查看文件我们可以知道,有686对蛋白找不到对应的MR和COR值
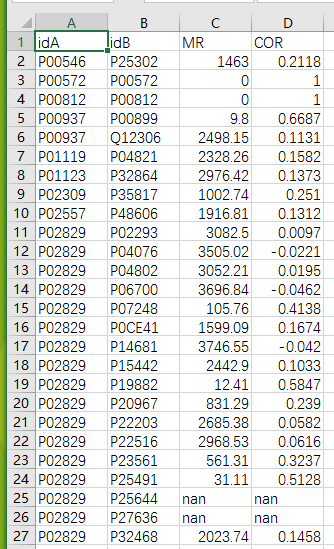
总结
Ensemble learning prediction of protein–protein interactions using proteins functional annotations
就像上面那篇论文所说的一样,缺失值太多,不适合使用,3006个样本有686 个缺失值,将近缺少了四分之一的值。
©著作权归作者所有:来自ZhiKuGroup博客作者没文化的原创作品,如需转载,请注明出处,否则将追究法律责任
来源:ZhiKuGroup博客,欢迎分享。


评论专区