
顺式调节图案
转录调控
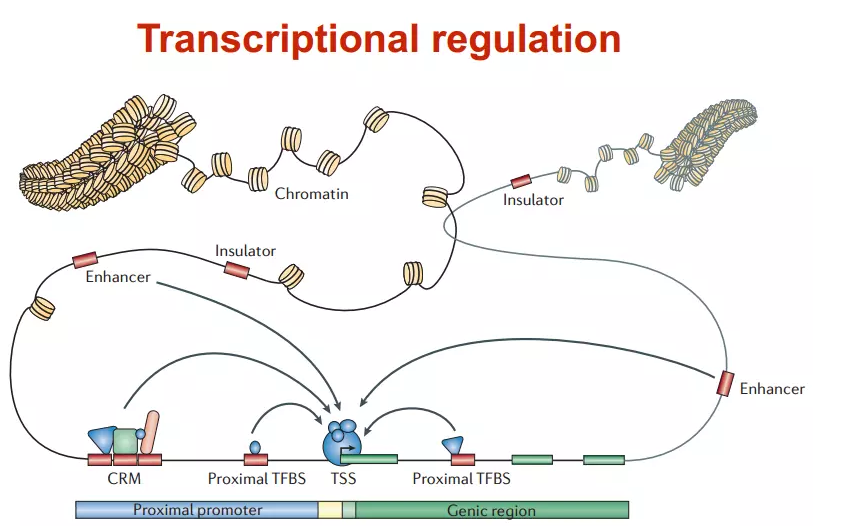
转录起始位点(TSS)
转录因子结合位点(TFBS)
顺式调节模块(CRM)有多个TF在一起
近端启动子和远端增强子近端的启动子远端的增强子
在人中,有300个TF结合在核心启动子区域;有1500个结合在基因其他区域,可以调节一系列基因
图示
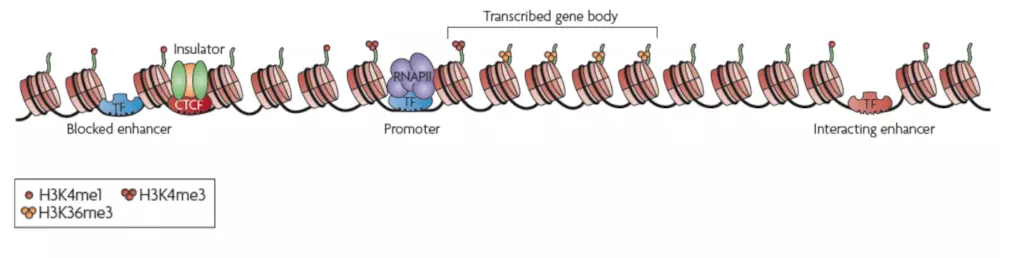
其中的绝缘体可以阻隔增强剂起作用
全基因组研究调控原件的主要方法
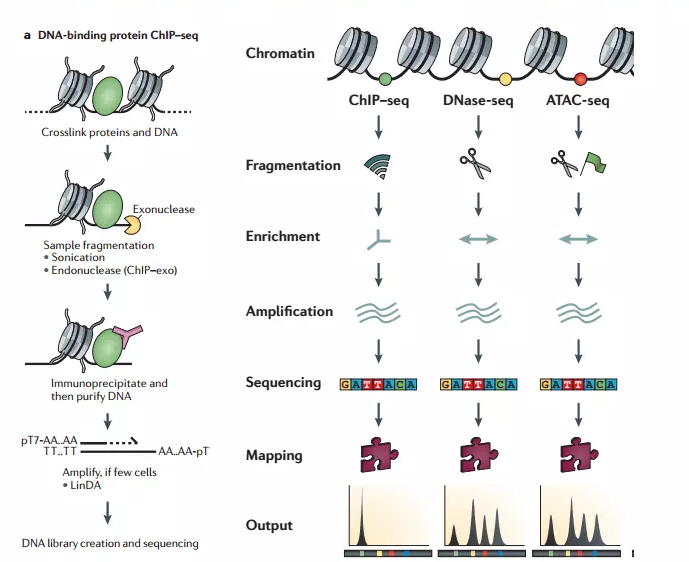
芯片起:
峰值的高低体现了蛋白的富集程度
只能研究单一的蛋白,具有特异性
有时候不能找到合适的抗体
DNA酶-SEQ
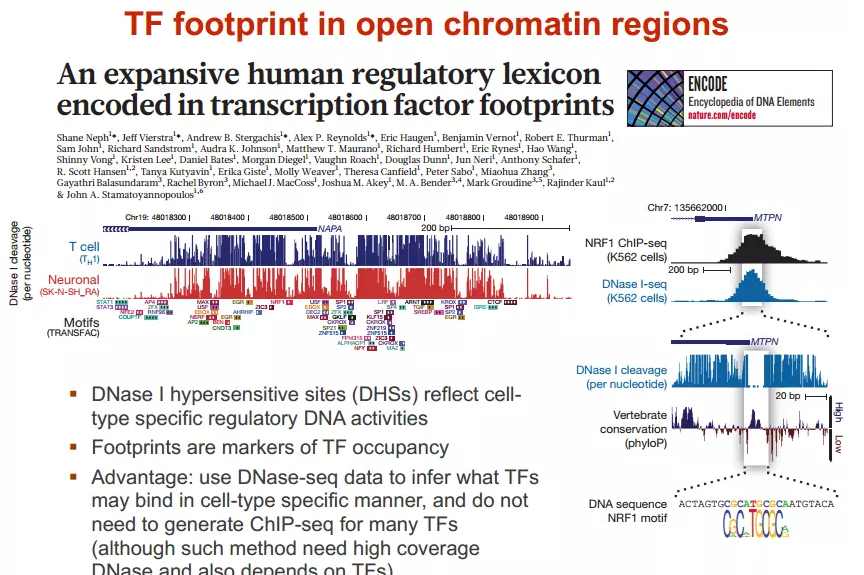
DNase I超敏感位点(DHS)是对DNaseⅠ高度敏感的活性染色质区域,DNase测序(DNase-seq)是进行全基因组DHS分析的常用方法
DNase I是一种非特异性核酸内切酶,基于它们对切割的过敏性,长期以来就被用于对“开放”染色质位点的作图
染色质打开的位置很容易有其他蛋白的结合
由于多种蛋白质可以与相同序列相结合,有必要整合DNA酶的SEQ测序数据和芯片起测序数据来对引起特定DNA酶足迹的蛋白质进行定性鉴定
不依赖于抗体或表位标签,DNase的序列可以用来在一次实验中分析大量蛋白质的基因组分布
从大范围来看,结合的位置凸起。如果从小范围来看,空着的位置刚好是一个可能的主题
ATAC-seq(使用测序检测转座酶 - 可及的染色质)
通过的Tn5转座酶,优先标记和测序核小体之间的DNA
ATAC-SEQ提供的信息与新的DNase-seq的法差不多,但步骤更为简单,需要的细胞也更少
在无法获得大量细胞的情况下,ATAC-SEQ更有帮助。
文章原图
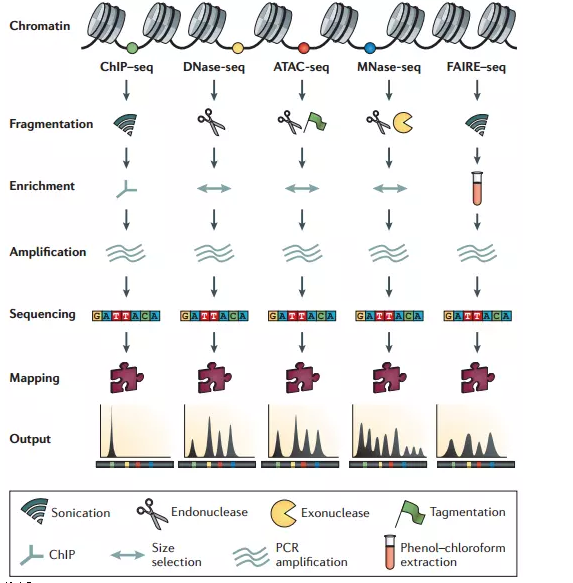
通过互补染色质配置实验分析的基因组基因座揭示了染色质结构的不同方面
ChIP-seq揭示了特异性转录因子(TF)的结合位点; DNase-seq,ATAC-seq和FAIRE-seq显示开放染色质区域; 和MNase-seq识别定位良好的核小体。
在ChIP-seq中,特异性抗体用于直接或通过含有靶因子的复合物中的其他蛋白质提取与靶蛋白结合的DNA片段。
在DNase-seq中,染色质被DNA酶I内切核酸酶轻微消化。大小选择用于富集在染色体区域中产生的片段,其中DNA对DNaseI攻击高度敏感。
ATAC-seq是DNase-seq的替代方法,其使用工程化的Tn5转座酶切割DNA并将引物DNA序列整合到切割的基因组DNA中(即标记)。
微球菌核酸酶(MNase)是内切核酸外切酶,其逐步消化DNA直至达到阻塞物,例如核小体。
在FAIRE-seq中,甲醛用于交联染色质,苯酚 - 氯仿用于分离剪切的DNA。
结合在哪里
一些TF几乎总是在近端启动子区域结合
其他人则绑定到许多地区
常用的表示方法
位置权重矩阵(PWM)
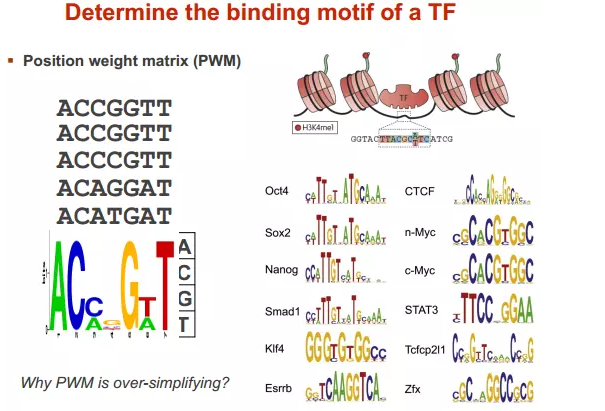
把所有碱基出现的次数相加,高度表示可信度
这种方法过于简单,不能表示出碱基之间的关系。
假设各个碱基之间均为独立
如何实现结合的特异性
主题定义

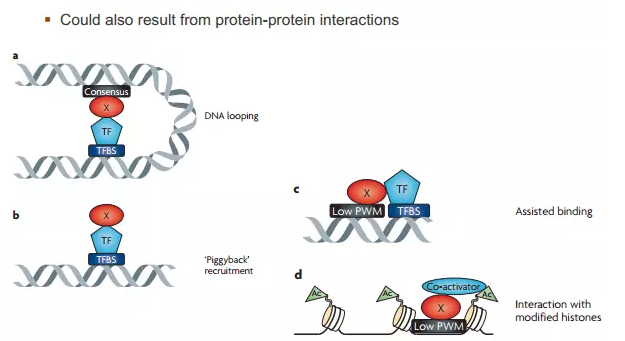
有时并非直接和DNA结合
IMG
如何识别TF绑定站点?

没有序列比对的时候
鉴于可能受相同TF调节的基因集合(或跨不同物种的直系同源基因 - 基于系统发育足迹原理的方法),找到共同的TF结合基序
但是问题是不知道主题是什么,找不到相关的基因,而且如何排除背景干扰
最原始的方法是多重序列比对MSA
比较保守的非编码区域可能有
PhyloCon - 比较基因组方法
结合序列比对和共表达基因
共表达基因很可能收到相同的主题调控
但并不是所有的元素都保守
Expectation-Maximization(EM)目前最常用的方法(MEME)
期望最大化
在每次迭代中,它学习PWM模型并识别矩阵的示例(输入序列中的站点)在每一次迭代中,学习一个PWMmodel然后再通过输入的序列进行比对
MEME通过迭代优化PWM并识别每个PWM的站点来工作(不同的迭代直到找到一个最合适的PWM)
直观的想法如下:
估计主题模型(PWM)
以k-mer种子(随机或指定)开始通常是6个
通过结合一些背景频率构建PWM根据背景生成一个初始的PWM
确定模型的示例
对于输入序列中的每个k聚体,在给定PWM模型的情况下识别其概率。计算k-mer在输入序列中给出PWM出现的概率
重新估计主题模型
根据输入序列中所有k-mers的加权频率计算新的PWM 根据输入序列中k-mer出现频率的权重更新PWM
迭代优化PWM并识别站点直到收敛
例子1

1.1

1.2

1.3

在MEME中
相关文献

过程
首先设置模型,然后经历Estep和Mstep,找到合适的PWM
然后将PWM进行极大似然转换并取日志

然后看输入序列中出现该主题的概率

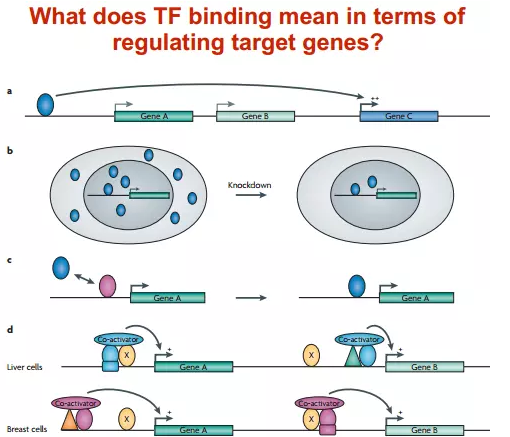
转录因子结合在调节靶基因方面意味着什么?
人的大多数结合位点都是在内含子和基因间区
更强的位点不接近差异调节的基因(不一定更具功能性)
大多数功能性站点未保存
目前很难预测靶基因
ChIP-seq调用峰值的策略和思想
的ChIP-seq的技术
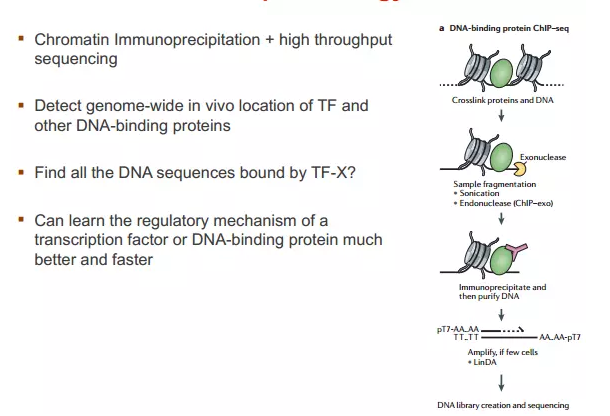
核心思想
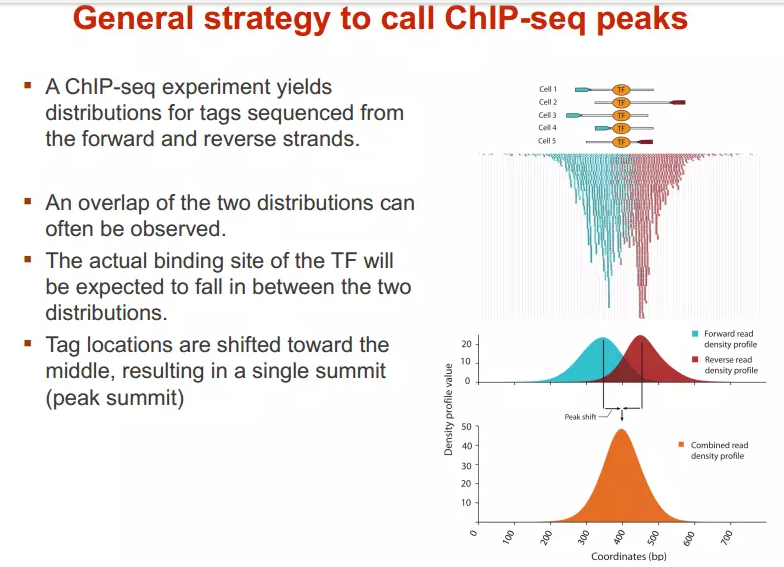
淅淅沥沥的原理
TF在基因组上的结合其实是一个随机过程,基因组的每个位置其实都有机会结合某个TF,只是概率不一样
峰值出现的位置,是TF结合的热点,而峰值通话就是为了找到这些热点。
热点:位置多次被测得的读所覆盖(我们测的是一个细胞群体,读出现次数多,说明该位置被TF结合的几率大)。
阅读出现多少次算多:假设TF在基因组上的分布没有任何规律,测序得到的阅读在基因组上的分布也必然是随机的,某个碱基上覆盖的阅读的数目应该服从二项分布。
当Ñ很大,对很小时,二项分布可以近似用泊松分布替代
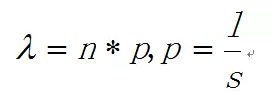
\拉姆达是泊松分布唯一的参数中,n是测序得到的读总数目,L是单个读的长度,S是基因组的大小。
我们可以算出在某个置信概率(如0.00001)下,随机情况下,某个碱基上可以覆盖的读取的数目的最小值,当实际观察到的读出数目超过这个值(单侧检验)时,我们认为该碱基是TF的一个结合热点。反过来,针对每一个读数目,我们也可以算出对应的置信概率P.
实际情况由于测序,映射过程内在的偏好性,以及不同染色质间的差异性,相比全基因组,某些碱基可能内在地会被更多的读取所覆盖,这种情况得到的很多峰可能都是假的。
MACS考虑到了这一点,当对某个碱基进行假设检验时,MACS只考虑该碱基附近的染色质区段(如10K),此时,上述公式中Ñ表示附近10k的区间内的读取数目,小号被置为10K。当有对照组实验(对照,相比实验组,没有用抗体捕获TF,或用了一个通用抗体)存在时,利用控制组的数据构建泊松分布,当没有控制时,利用实验组,稍大一点的局部区间(比如50K)的数据构建泊松分布。
读只是跟随着TF一起沉淀下来的DNA片段的末端,读的位置并不是真实的TF结合的位置。
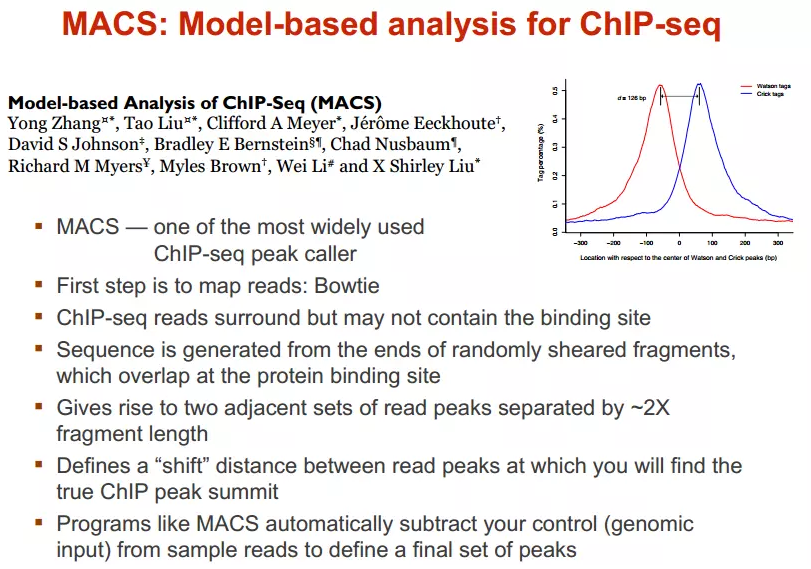
在峰打电话之前,延伸阅读是必须的。不同TF大小不一样,对阅读延伸的长度也理应不同。
我们知道测得的读取最终其实会近似地平均分配到正负链上,这样对于一个TF结合热点而言,阅读在附近正负链上会近似地形成“双峰”。
MACS会以某个窗口大小扫描基因组,统计每个窗口里面读的富集程度,然后抽取(比如1000个)合适的(读富集程度适中,过少,无法建立模型,过大,可能反映的只是某种偏好性)窗口作样本,建立“双峰模型”。
最后,两个峰之间的距离就被认为是TF的长度d,每个读将延伸d / 2的长度
的ChIP-seq的后续分析
如果给出一组ChIP-seq峰,如何识别TF-使用MEME的基序
要找出序列主题类似的东西 - 使用TomTom
使用已知的主题搜索峰值区域 - 使用FIMO
研究TF的潜在靶基因的常见生物途径或功能 - 使用GREAT
刘晓乐实验室的ChIP-seq的数据分析流程
基因调控网络
贝叶斯网络
定义:包括一个有向无环图(DAG)和一个条件概率表集合.DAG中每一个节点表示一个随机变量,可以是可直接观测变量或隐藏变量,而有向边表示随机变量间的条件依赖;条件概率表中的每一个元素对应的DAG中唯一的节点,存储此节点对于其所有直接前驱节点的联合条件概率
性质:每一个节点在其直接前驱节点的值制定后,这个节点条件独立于其所有非直接前驱前辈节点
类似的马尔可夫过程,贝叶斯网络可以看做是马尔可夫链的非线性扩展。这条特性的重要意义在于明确了贝叶斯网络可以方便计算联合概率分布。
通过基因表达来推测网络
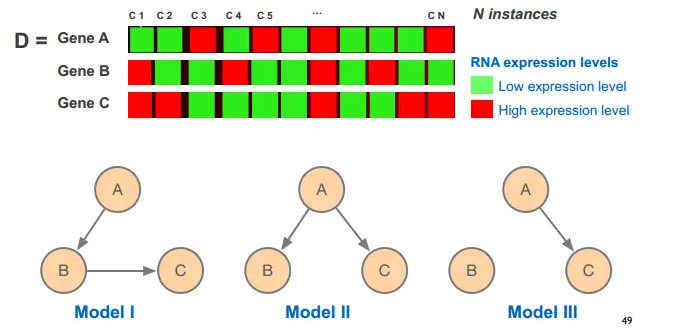
模型图形说明
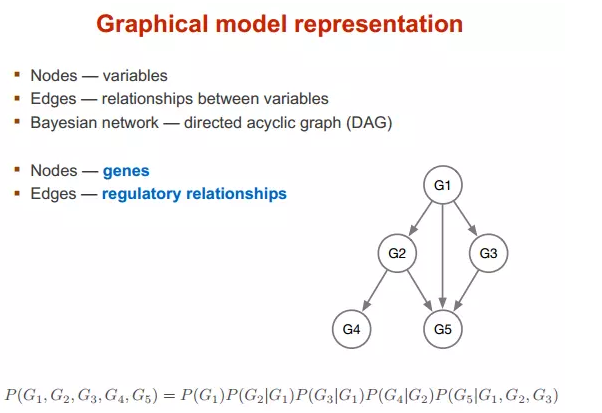
DAG:有向无环图
条件概率分布(CPD)条件概率分布
多变量非独立联合条件概率分布P(G1,G2,G3,G4,G5)求取公式
模型选择
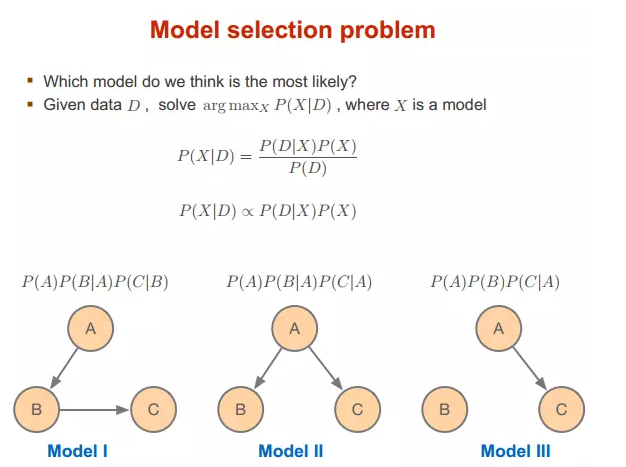
作者:思考问题的熊
链接:https://www.jianshu.com/p/7f202984803d
來源:简书
简书著作权归作者所有,任何形式的转载都请联系作者获得授权并注明出处。


评论专区