
NCBI数据库文件扩展名解析*.1
For more details, contact the NCBI help desk: info@ncbi.nlm.nih.gov .asn genome record in asn.1 format .faa protein sequences in fasta format, text file .ffn protein coding portions of the genome segments .fna genome fasta sequence .frn rna coding portions of the genome segments .gbk genome in genbank file format .gff genome features .ptt protein table .rnt rna table .rpt summary report .val binary file (genome project?) Other extensions, and my understanding of their meaning: .gb Genbank? .gpff Genbank protein
*.1 文件结尾
ASN.1格式 — 摘要句法记号1,国际标准组织(ISO)数据表示格式,下载大多数最近的完全公告和日常积累或非积累更新数据。
FASTA格式 — 定义行号后只跟随序列数据(示例),参见描述数据库的readme文件,包括nt.Z(每天更新的非冗余BLAST核酸数据库,包括GenBank+EMBL+DDBJ+PDB序列,但是不包括EST, STS, GSS, or HTGS序列),nr.Z(每日更新的非冗余蛋白质),est.Z, gss.Z, htg.Z, sts.Z,和其它文件。
使用别人发表的高通量测序数据进行挖掘,筛选差异基因进行后续研究,不仅节省经费,又节约时间,可以大大提升研究人员的工作效率。今天小编就给大家介绍下如何从NCBI数据库下载高通量测序数据。
1、进入NCBI SRA数据库搜索项目界面,输入SRP编号。SRP编号一般可以文章中找到。现在大部分杂志都需要作者上传测序数据到公共数据库,并将提交得到的编号SRP编号(测序)或GSE编号(芯片)放到文章中。
https://www.ncbi.nlm.nih.gov/Traces/study/
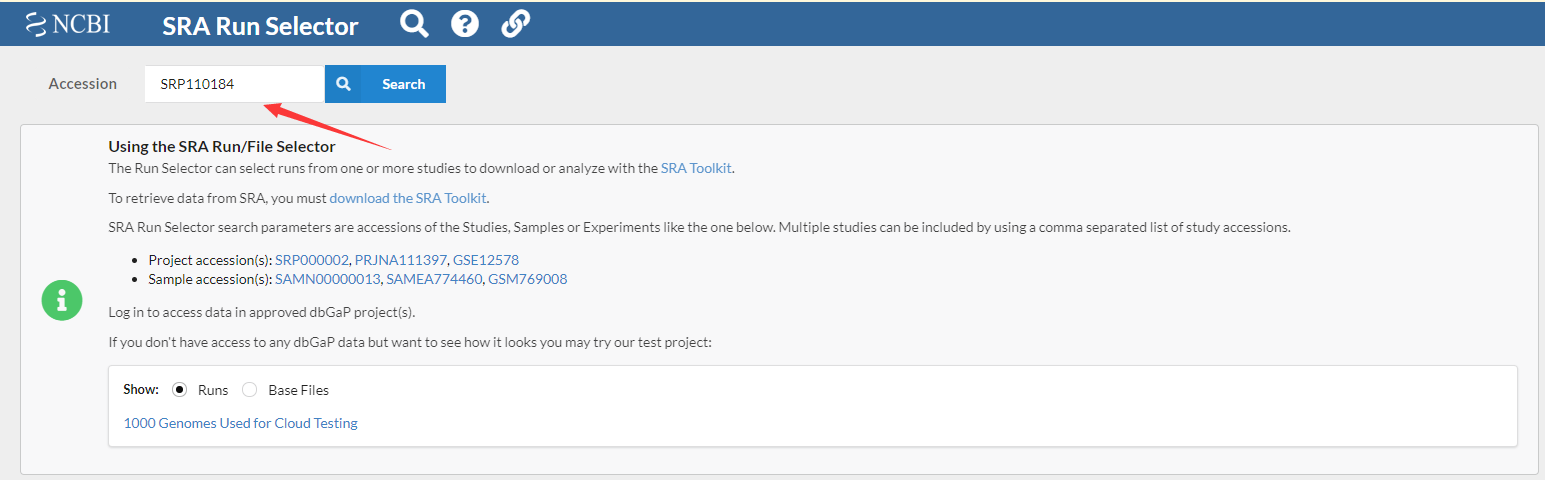
2、输入SRP编号后出来界面会显示测序数据信息,我们输入SRP110184点击搜索后,会找到8个样本的测序数据。
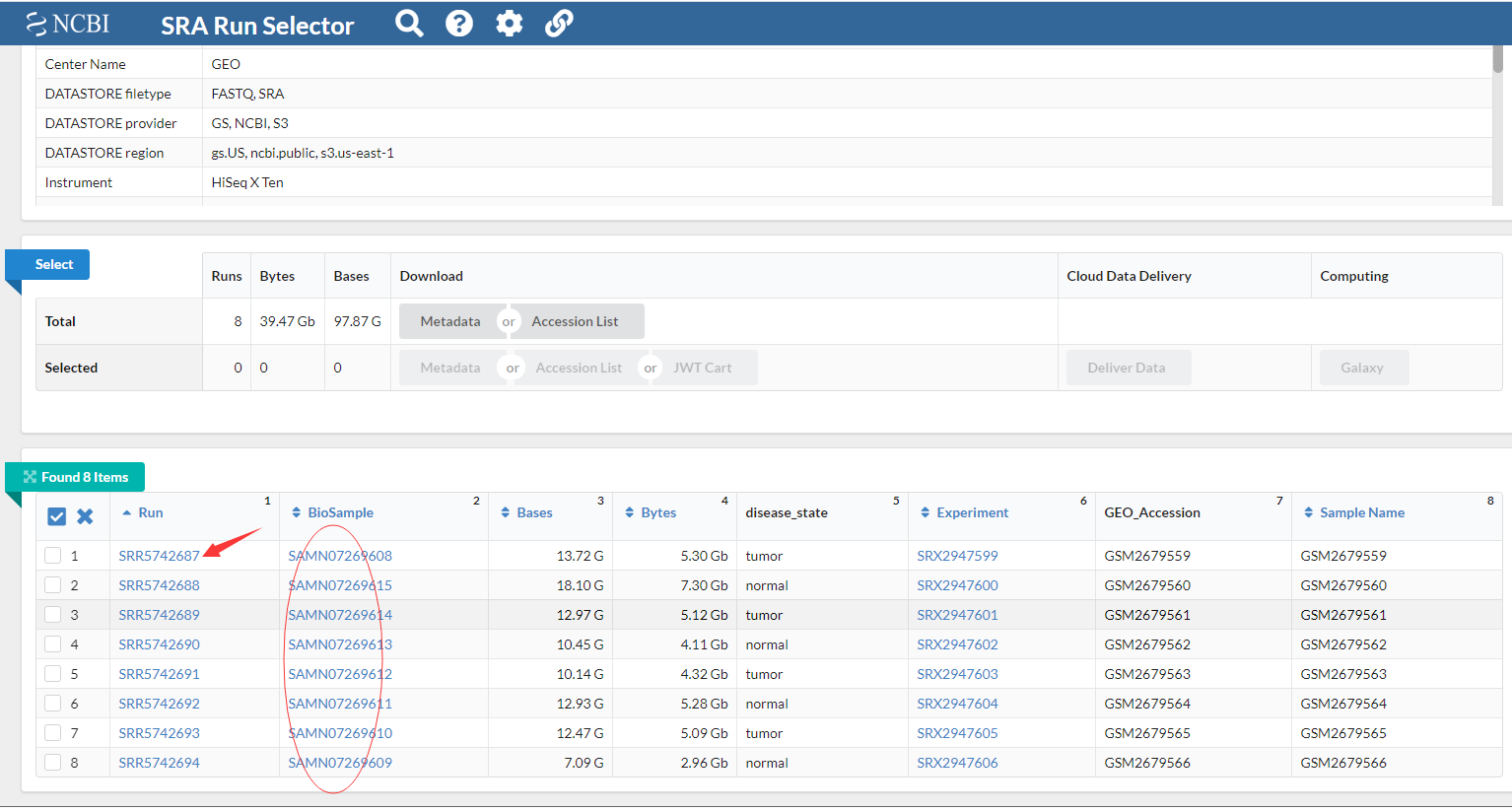
3、数据下载,第一可以直接点击单个样本,进行页面下载。Data access界面有两个下载链接都可以下载。
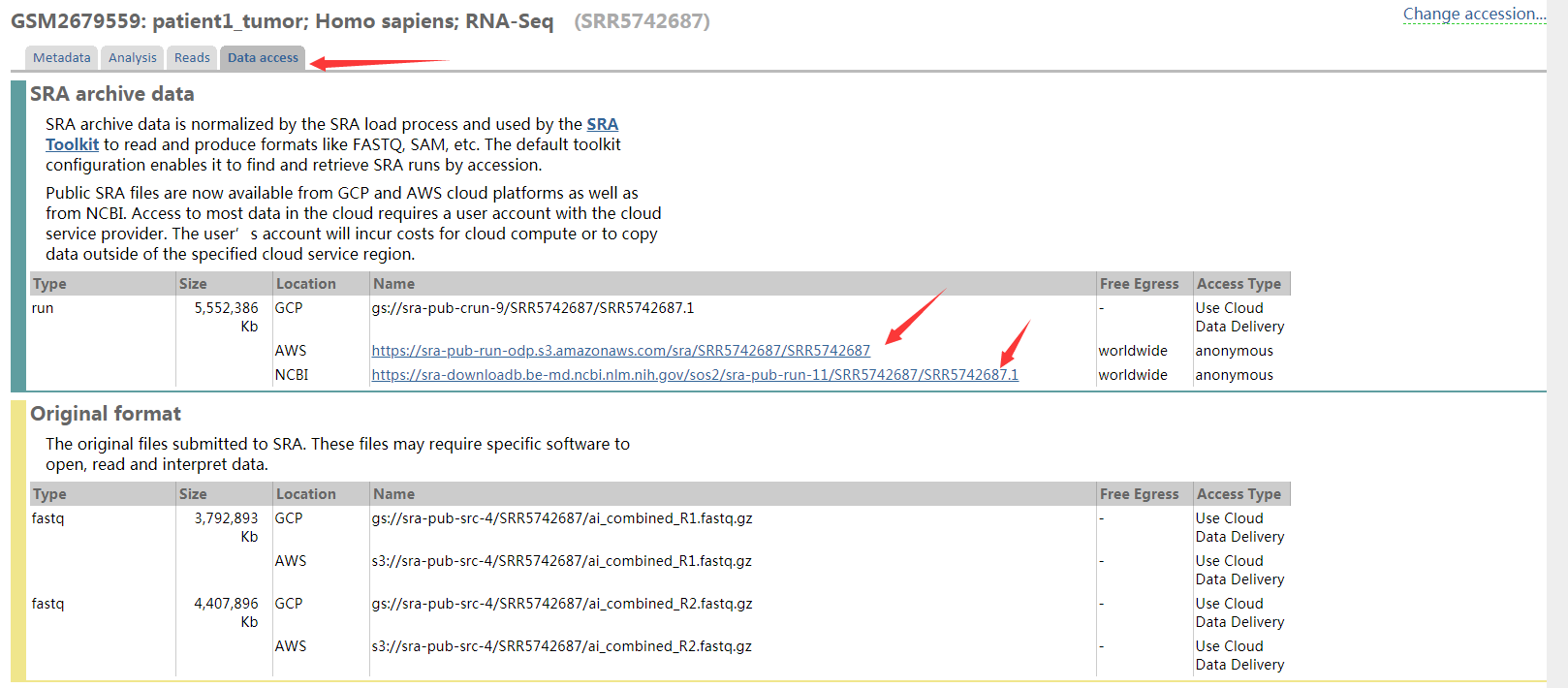
4、下载的数据需要转化为fastq格式才能进行下一步的差异表达分析。这里就需要NCBI提供的下载工具SRA Toolkit进行转化。AWS,NCBI两个链接下载的文件后缀为”.man”,”.1”格式。
https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software

5、根据操作系统选择相应版本的软件下载。Window版本软件下载后为一个压缩文件 sratoolkit.2.11.0-win64.zip。解压缩后,设置环境变量后就可以使用。
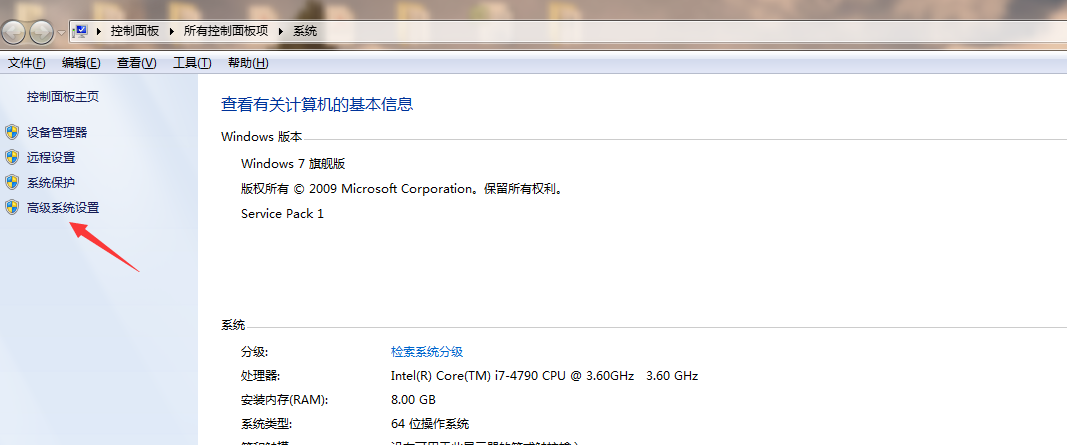
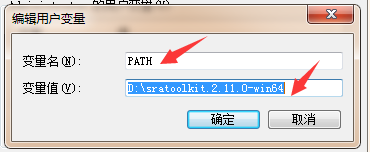
具体安装方法:鼠标移至“计算机”图标,点击右键---属性---高级系统设置---环境变量---PATH---新建---输入sratoolkit.2.11.0-win64解压缩所在的路径,点击确认就可以使用了。

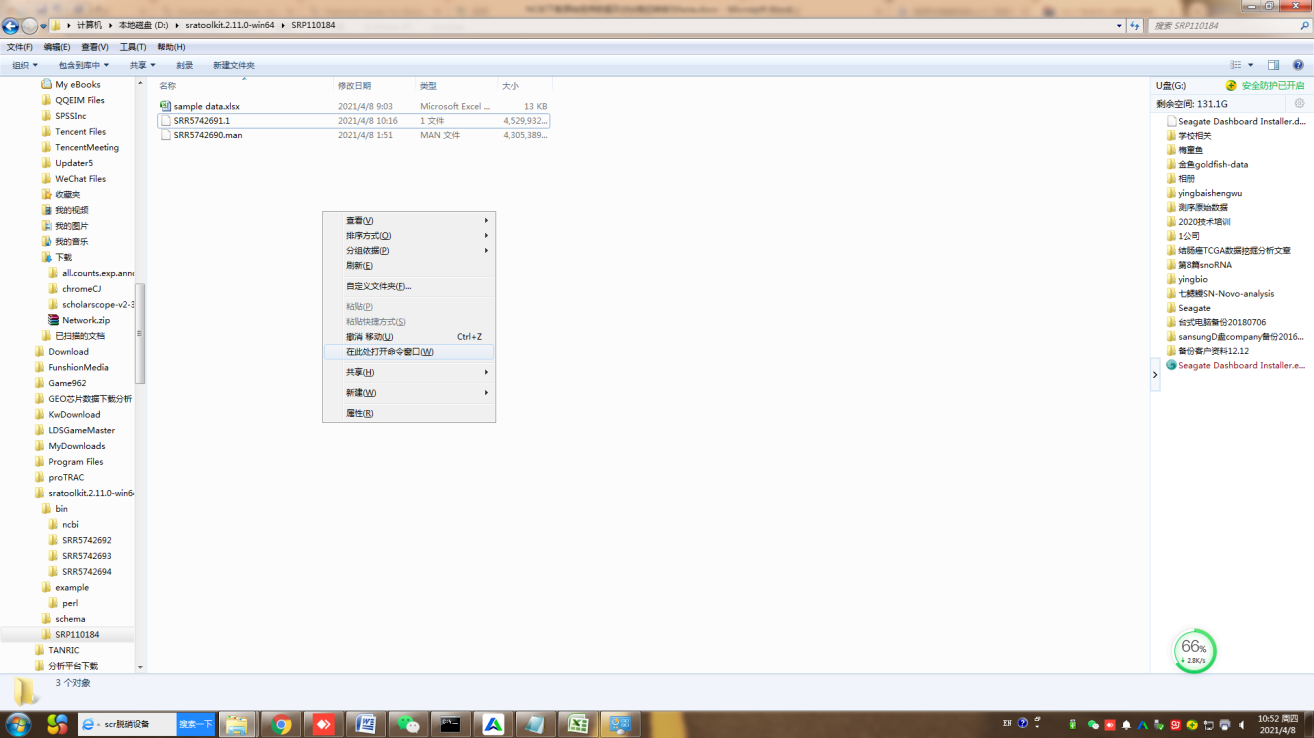
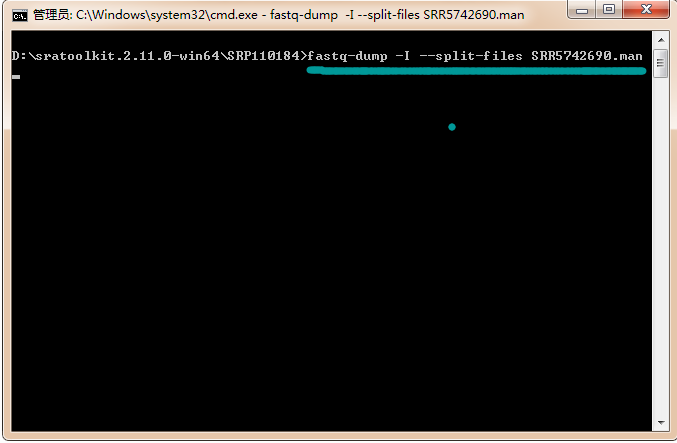
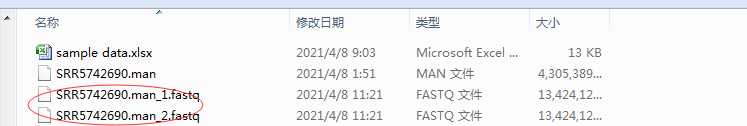
6、SRA下载文件转化为fastq格式文件 。在测序下载文件中,按shift键同时点击右键,在出来的下拉框中点击“在此处打开命令框”,出现dos界面。输入命令fastq-dump -I --split-files SRR5742690.man, SRR5742690.man为下载数据文件名及后缀。程序运行完成后会两个fastq文件,是测序数据的两个双端文件。
7、如果样本比较多可以通过NCBI提供的下载工具SRA Toolkit下载。在SRA Run Selector界面下载所有样本信息Accession list(下载得到SRR_Acc_List.txt文件)。
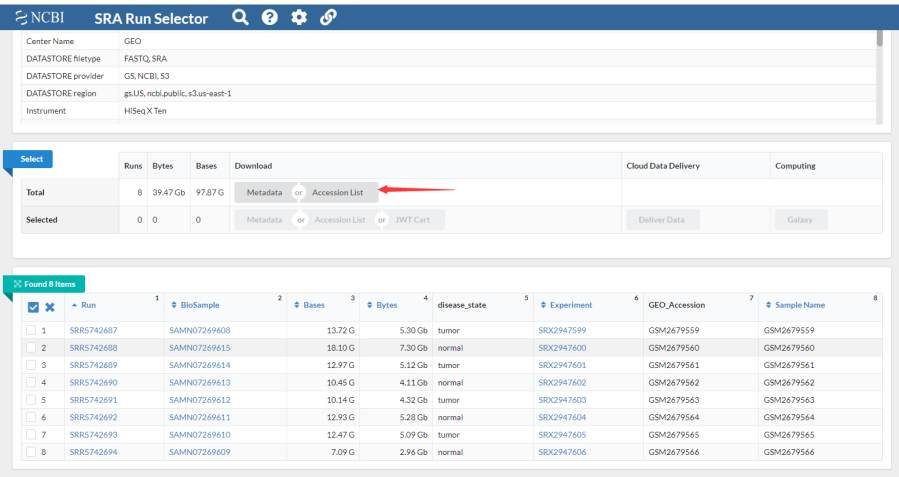
8、将RR_Acc_List.txt放到sratoolkit.2.11.0-win64文件夹中bin文件夹中,在bin文件夹中按住shift同时点击邮件打开dos命令框,输入命令:prefetch.exe --option-file SRR_Acc_List.txt。程序会逐个下载各个样本的数据,一个样本一个文件夹。
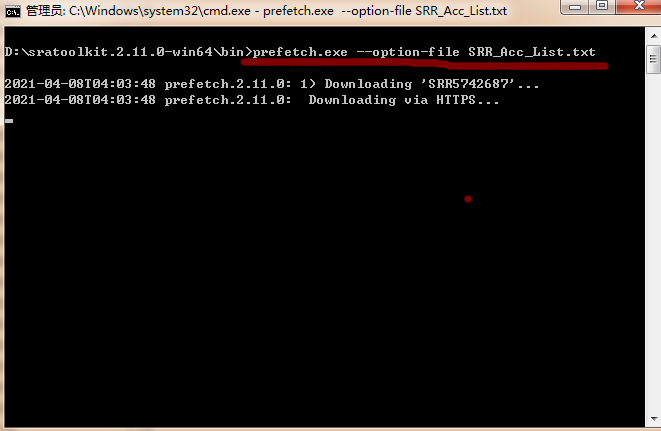
9、得到fastq双端测序数据之后就可以进行后续差异筛选,功能富集等分析了。
下载的数据需要转化为fastq格式才能进行下一步的差异表达分析。这里就需要NCBI提供的下载工具SRA Toolkit进行转化。AWS,NCBI两个链接下载的文件后缀为”.man”,”.1”格式。






评论专区